A Multi-Center Trial to Evaluate the Safety and Toxicity of Nanoxel®-M in Breast Cancer Patients
Article information
Abstract
Purpose
Nanoxel®-M is a low-molecular-weight, non-toxic, biodegradable, docetaxel-loaded methoxy-poly (ethylene glycol)-block-poly (D,L-lactide) (mPEG-PDLLA) micellar formulation. We conducted a multicenter trial to evaluate the safety and toxicity of Nanoxel®-M and the quality of life (QoL) of Korean breast cancer patients treated with this formulation.
Methods
Patients received adjuvant Nanoxel®-M with a schedule comprising four alternating cycles of doxorubicin with cyclophosphamide, followed by either Nanoxel®-M or Nanoxel®-M with cyclophosphamide after surgery for early breast cancer. We analyzed hematological and non-hematological toxicity profiles and alterations in patient QoL using the Korean version of the European organization for research and treatment of cancer core 30-item quality of life questionnaire. Fifty-five operable breast cancer patients with stage II or III disease were enrolled from four centers in Korea.
Results
Regarding safety and toxicity profiles, grade 3/4 toxicity presented as anemia in 0.5%, neutropenia in 61.8%, febrile neutropenia in 4.5%, mucositis in 1.4%, and edema in 0.5% of patients during 220 total cycles. However, all-grade thrombocytopenia was not observed among hematological toxicities. No grade 3/4 nausea, vomiting, diarrhea, hand foot syndrome, dyspnea, allergic reaction, edema, or peripheral neuropathy were observed. Furthermore, no vehicle-related hypersensitivity reactions occurred when using Nanoxel®-M.
Conclusion
Our findings indicate that Nanoxel®-M could be used to treat operable breast cancer patients, compare favorably with docetaxel in terms of hypersensitivity reactions and the incidence of taxane-induced peripheral neuropathy, and is associated with a similar incidence of febrile neutropenia.
INTRODUCTION
Docetaxel is a cytotoxic, taxoid, anticancer drug derived from 10-deacetylbaccatin III, which was isolated from extracts of the Western yew tree (Taxus baccata) in the 1970s [1]. Taxanes including docetaxel and paclitaxel, are mitotic inhibitors that bind to tubulin and stabilize GDP-bound tubulin in microtubules, thereby activating the polymerization of tubulin and inhibiting the depolymerization of microtubules during cancer cell mitosis. Consequently, these drugs induce cell division arrest and apoptosis in cancer cells [2-4]. Many studies have consistently shown that docetaxel has anticancer activity in preclinical and clinical settings. Currently, this drug is widely prescribed for the treatment of various types of cancer, including breast, lung, prostate, ovarian, esophageal, and gastric cancers in adjuvant or metastatic settings [5-7]. However, taxane compounds are characterized by poor water solubility; therefore, a formulation vehicle used for poorly water-soluble drugs, such as Chremophor EL (CrEL) or Tween 80, may be necessary [8,9].
Early in the clinical development of docetaxel, it was established that this drug is associated with adverse side effects, including acute hypersensitivity reactions and cumulative fluid retention caused by the amphiphilic solvent system, polysorbate-80 [10]. Researchers have focused on developing less-toxic, better-tolerated, polysorbate-free formulations based on the use of liposomes, micelles, macromolecular conjugates, submicron emulsions, prodrugs, and nanoparticles. Among these drug delivery systems, polymeric micelles have been investigated extensively for their drug-loading capacity and ability to deliver taxanes to solid tumors [11,12]. Nanoxel®-M (Samyang Biopharm. Co. Seongnam, Korea) was developed using amphiphilic, deblock, copolymer (mPEG-PDLLA)-based micelles for the solubilization of docetaxel, in an effort to avoid the toxic effects of the nonionic surfactant, polysorbate 80 (Tween 80), which is used in the commercial formulation of Taxotere®. With regards to vehicle-associated toxicity, investigators previously found significant differences between Nanoxel®-M and Taxotere® in terms of hypersensitivity reactions [13]. Accordingly, we planned a trial designed to obtain data regarding the safety and toxicity associated with Nanoxel®-M treatment in an adjuvant setting for breast cancer. We also sought to evaluate the alterations in patients’ quality of life (QoL) related to Nanoxel®-M treatment.
METHODS
This study was designed as a multicenter phase IV trial to determine the safety and toxicity profiles of Nanoxel®-M in Korean patients with breast cancer. We screened eligible patients with operable early- or locally-advanced breast cancer who were to be treated with a docetaxel-based regimen, which involved the alternating administration of doxorubicin with cyclophosphamide (AC) followed by either docetaxel or docetaxel with cyclophosphamide (TC) immediately after surgery or just prior to administration of the first cycle of docetaxel treatment.
The primary endpoint of our study was the assessment of safety and toxicity; the secondary endpoint was QoL. We analyzed toxicity and safety according to the National Cancer Institute’s common terminology criteria for adverse events (CTCAE, ver. 3.0), as well as QoL using a validated Korean version [14] of the European organization for research and treatment of cancer (EORTC) core 30-item quality of life questionnaire (QLQ-C30) version 3.0 [15]. The QLQ-C30 is composed of both multi- and single-item scales. These include five functional scales, three symptom scales, a global health status/QoL scale, and six single items. Each of the multi-item scales includes a different set of items, with no single item occurring in more than one scale. For scoring QoL, we adopted a procedure set out in a manual from the quality of life unit of the EORTC data center. When using this procedure, a high score for a functional scale is indicative of a healthy level of functioning; a high score for global health status equates to a high QoL. In contrast, a high score on the symptom scale indicates a high level of symptomatology or problems. The patients were interviewed at the time of baseline screening and during subsequent visits for chemotherapy at 3-weekly intervals.
Patient selection and baseline data
For the enrollment of patients in this study, we issued investigators with specific guidelines based on four molecular subtypes: luminal A, luminal B, human epidermal growth factor receptor 2 (HER2)-enriched, and triple negative breast cancer (TNBC). The luminal A subtype is defined as estrogen receptor (ER) and/or progesterone receptor (PR)-positive, HER2-negative, and low Ki67 (≤ 14%); the luminal B subtype is defined as ER- and/or PR-positive, HER2-negative and high Ki67 (>14%), or as ER- and/or PR-positive and HER2-positive; the HER2-enriched subtype is defined as ER- and PR-negative and HER2-positive; the TNBC subtype is defined as ER- and PR-negative and HER2-negative. A regimen involving the administration of AC followed by Nanoxel®-M was recommended for luminal B, HER-2 enriched, and TNBC subtype patients who were classified as high-risk patients (Group 1), whereas a Nanoxel®-M with cyclophosphamide regimen was recommended for luminal A subtype patients who were classified as low-risk (Group 2) (Figure 1). However, we permitted the participating investigators to select the regimen they eventually adopted based on personal preference, because the primary aim of our study was to determine the safety and toxicity profiles of Nanoxel®-M.

Study design of adjuvant Nanoxel®-M. AC = adriamycin+cyclophosphamide; TC = docetaxel (Nanoxel®-M)+cyclophosphamide; T = docetaxel (Nanoxel®-M); HER2 = human epidermal growth factor receptor 2; ER = estrogen receptor; PR = progesterone receptor.
To be eligible for this study, patients needed to have histologically or cytologically confirmed invasive breast cancer with metastasis to ipsilateral axillary lymph nodes or a tumor larger than 2 cm. Further eligibility criteria included female sex, age between 20 and 70 years, Eastern cooperative oncology group (ECOG) performance status between 0 and 2 with a life expectancy of more than 6 months, adequate hematological [hemoglobin ≥ 10 g/dL, absolute neutrophil count (ANC) ≥ 1,500/mm3, platelet count ≥ 100,000/mm3], renal (serum creatinine ≤ 1.5 mg/dL), hepatic (total bilirubin ≤ 1.5 mg/dL, transaminases and alkaline phosphatase ≤ 3× the upper normal limit), and cardiac (normal electrocardiography [EKG] within a months or left ventricular ejection fraction [LVEF] >50% within 3 months) functions.
We excluded patients with a second primary malignancy (with the exception of carcinoma in situ in the cervix, adequately treated non-melanoma skin cancer, and thyroid cancer), peripheral neuropathy ≥ grade 2, an uncontrolled infectious condition, a psychiatric disease, an epileptic disorder, ventricular or atrial arrhythmia, congestive heart failure, cardiac infarction, unstable angina, distant metastasis, or any serious concomitant systemic disorder.
After screening for enrollment, we obtained informed consent from all eligible patients and documented baseline data, including medical history (family history, menopause, concomitant disease, medication, allergy, and weight loss), physical examination (height, weight, body surface area, ECOG performance status, and neurological test), laboratory blood tests (complete blood counts [CBC] with differential counts) with biochemical analyses (calcium, phosphorus, glucose, blood urea nitrogen [BUN], creatinine, cholesterol, protein, albumin, and total bilirubin), tumor marker (CA15-3) assessment, urinalysis, and cardiopulmonary function tests (chest X-ray, electrocardiogram, echocardiography, or multigated acquisition [MUGA] scan). Baseline imaging examination requirements were as flexible as possible, and included any one of the following: mammography, breast ultrasound, bone scan or 18F-fluorodeoxyglucose–positron emission tomography (PET-CT), chest CT, or abdominal CT performed prior to surgery.
Initial treatment plan
Eligible patients received four cycles of Nanoxel®-M 100 mg/m2 as a 1-h intravenous (IV) infusion every 3 weeks after four cycles of doxorubicin 60 mg/m2 plus cyclophosphamide 600 mg/m2 (day 1) every 3 weeks (Group 1), or four cycles of Nanoxel®-M 75 mg/m2 plus cyclophosphamide 600 mg/m2 IV infusion every 3 weeks after surgery (Group 2). We routinely administered IV dexamethasone 10 mg, phenyramine maleate 45.5 mg, and ranitidine 50 mg or cimetidine 300 mg prior to each cycle of Nanoxel®-M to prevent potential hypersensitivity reactions to docetaxel treatment. Prophylactic use of colony-stimulating factors (filgrastim or granulocyte colony-stimulating factor [G-CSF]) for neutropenia was not permitted.
Change in treatment plan
From the commencement of this study, we observed a higher-than-expected incidence of febrile neutropenia in Group 1 patients who received a 100 mg/m2 dose of Nanoxel®-M. All participating investigators agreed to modify the treatment plan by using 75 mg/m2 of Nanoxel®-M, instead of 100 mg/m2, for the treatment of Group 1 patients. Consequently, we had two different dosage subgroups (100 mg/ m2 and 75 mg/m2 Nanoxel®-M) in Group 1, and thus three different analysis groups: Group 1 (100), Group 1 (75), and Group 2.
Dose modification
In this study, we used the National Cancer Institute common toxicity criteria (NCI-CTC) grading system for toxicity assessments. The doses of the study drugs were interrupted or modified for grade 3-4 hematological toxicities and grade 3-4 non-hematological toxicities, according to the protocol. For hematological toxicities, dose resumption could be delayed for a maximum of 3 weeks and commenced only for patients with ANC ≥ 1,500/mm3 and platelet counts ≥ 100,000/mm3. Dose reductions of 25% were mandated for patients who experienced grade 3 or 4 neutropenia associated with a fever of >38.5°C and were maintained until the next cycle of chemotherapy, even if patients had regained the normal range of CBC. Only two dose reductions were permitted; for patients who experienced febrile neutropenia, the doses of all study drugs were reduced by 25% in all subsequent cycles. Patients requiring more than two further dose reductions were withdrawn from the study. For all grade 3 or higher non-hematological toxicities, treatment was delayed until the patient had recovered to grade 1 or less. Patients with grade 4 hypersensitivity reactions were withdrawn from the study.
Study assessments
The primary end point of this study was the analysis of the safety and toxicity profiles of patients within the study population who had received at least one dose of Nanoxel®-M. We graded adverse events for toxicity and safety profiles according to the CTCAE at 3-weekly intervals based on physical examination and laboratory tests. The secondary end point of this study was the evaluation of the alterations in the patients’ QoL index from the initial baseline prior to initiating Nanoxel®-M treatment until to the end of the examined treatment period. The scoring procedure we used was based on The EORTC QLQ-C30 scoring manual (3rd edition), published by the European organization for research and treatment of cancer in Brussels (2001). Intragroup comparisons were conducted for each treatment cycle; data were analyzed using repeated-measures ANOVA, with overall differences assessed using Friedman’s test. All statistical analyses were conducted using SPSS ver. 21 (IBM Corp., NY, USA), for which we adopted a two-sided significance level of 5%. We interpreted increases in the mean-transformed QLQ C30 physical functioning score (FS) and global health score (GHS) over time as improvements. In contrast, an increase in the mean-transformed QLQ C30 symptom score (SS) over time was interpreted as deterioration.
Compliance with ethical standards
All procedures were performed in accordance with the guidelines for Good Clinical Practice and the Declaration of Helsinki, and the study was approved by the institutional review board (IRB) of each hospital. The local IRB number of Inje University Ilsan Paik Hospital is 2014-06-222. All participants provided written informed consent prior to enrollment.
RESULTS
Patient characteristics
A total of 55 patients from four centers in Korea were enrolled in the study. The data for patients with operable breast cancer who received four cycles of Nanoxel®-M without dropping out were used to assess safety and toxicity. All patients were allocated into one of two study groups, namely Group 1 (AC followed by Nanoxel®-M) and Group 2 (TC), according to investigator’s guidelines. Forty-five patients were included in Group 1 and the remaining 10 patients were allocated to Group 2. As previously mentioned, we reduced the Nanoxel®-M dosage from 100 mg/m2 to 75 mg/m2 in Group 1 patients due to the high incidence of febrile neutropenia.
The characteristics of the study patients, including tumor size, nodal status, tumor-node-metastasis (TNM) stage, histological type, histological grade, hormonal receptor, HER2 expression, and Ki 67 index, are listed in Table 1. The median age of the study group was 55.9 years (range, 48.6–63.2 years), approximately one-third of whom were premenopausal and two-thirds were postmenopausal. In most cases (52 patients [94.5%]), the T stage was T1 or T2; in the majority of patients (41 patients [74.5%]), the N stage was N1. Among all histological types, invasive ductal carcinoma (IDC) presented in 49 patients (89.1%); histological grades (HGs) II and III were recorded for 45 patients (81.8%). Two-thirds of the enrolled patients had ER- and/or PR-positive tumors; one-third had HER2-positive tumors. Forty patients (72.7%) had a low Ki 67 index (less than 14%); the remaining 15 patients (27.3%) had intermediate or high Ki 67 indices (greater than 14%).
Demographic and clinicopathological characteristics of patients treated with Nanoxel®-M in different dosage groups
Toxicity profiles
We analyzed toxicity profiles of all grade and grade 3/4 adverse hematological and non-hematological events, associated with Nanoxel®-M treatment during all cycles. All grade hematological toxicities included anemia (35.0%), neutropenia (84.5%), and febrile neutropenia (4.5%), whereas no thrombocytopenia was reported. All grade non-hematological toxicities included mucositis in 6.8% of patients, nausea in 14.1%, vomiting in 0.9%, diarrhea in 3.2%, hand foot syndrome in 4.5%, dyspnea in 1.8%, allergic reaction in 0.9%, edema in 20.9%, and peripheral neuropathy in 9.1% (Table 2).

All grade toxicity profiles of patients treated with Nanoxel®-M in different dosage groups
Grade 3/4 adverse hematological events associated with Nanoxel®-M treatment during all cycles included anemia (0.5%), neutropenia (61.8%), and febrile neutropenia (4.5%). However, no instances of thrombocytopenia have been reported to date. Additionally, grade 3/4 adverse non-hematological events associated with Nanoxel®-M treatment during all cycles included mucositis in 1.4% of patients and edema in less than 1% (Table 3). Although we recorded frequent changes in the body weight of half of the study patients, grade 1 weight gain was recorded in only 11 patients, grade 2 weight gain in seven, and grade 1 weight loss in two patients.

Grade III or IV toxicity profiles of patients treated with Nanoxel®-M in different dosage groups
Following reduction of the Nanoxel®-M dose from 100 mg/m2 to 75 mg/m2 in Group 1 patients, we observed a decrease in the incidence of hematological and non-hematological toxicities. Neutropenia decreased from 90.3% to 81.5% in all grades and from 81.9% to 56.5% in grades 3/4; mucositis decreased from 11.1% to 6.5% in all grades and from 2.8% to 0.9% in grades 3/4; peripheral neuropathy decreased from 16.7% to 5.6% in all grades, although no grade 3/4 decreases were recorded (Tables 2, 3).
QoL
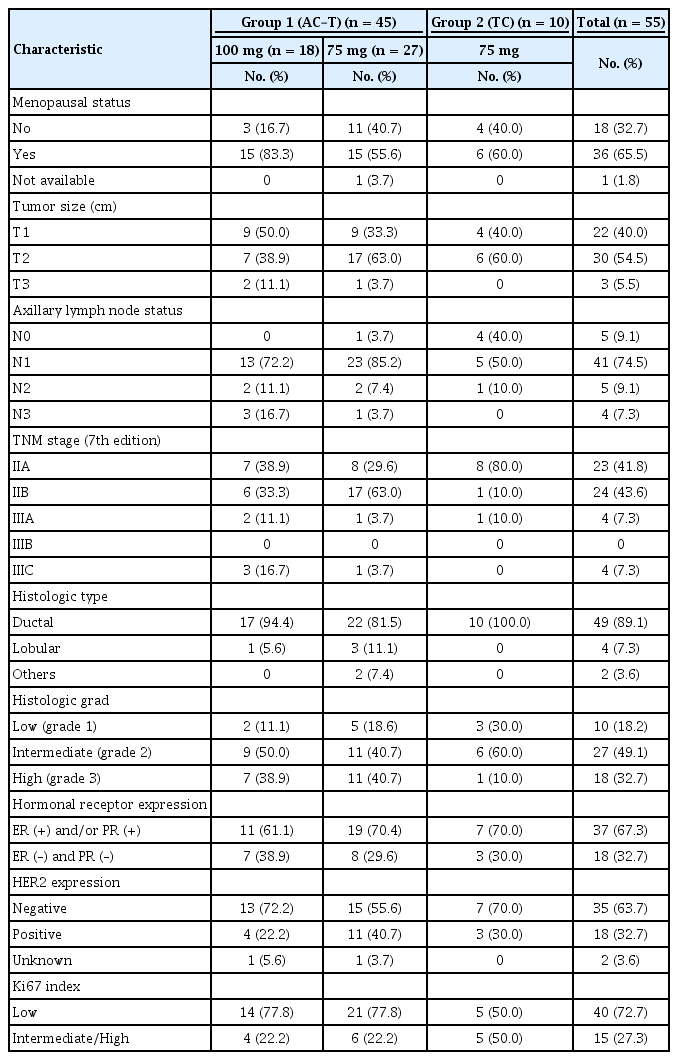
For enrolled breast cancer patients in the 100 mg dosage subgroup of Group 1, the EORTC QLQ-C30 FS decreased concomitantly with an increase in the number of treatment cycles, changing from 75.6 at baseline to 74.9, 71.7, 69.5, and 59.5 at cycles 1, 2, 3, and 4, respectively. In contrast, the FS of patients in the 75 mg dosage subgroup of Group 1 showed an initial increase followed by a decrease over time, from 66.4 at baseline to 75.4, 70.3, 64.9, and 63.4 at cycles 1, 2, 3, and 4, respectively. In Group 2 patients, the FS gradually decreased from 75.7 at cycle 1 to 75.6, 72.2, 64.9, and 69.1 at cycles 2, 3, and 4, and the end of the trial, respectively (Figure 2A). We found that the mean FS value was highest among Group 2 patients, whereas the difference between each FS value was largest in the 100 mg dosage subgroup of Group 1 and smallest in Group 2, which was statistically significant (p= 0.0370; Figure 2B).
EORTC QLQ-C30 functioning score (FS) of patients treated with Nanoxel®-M. (A) Changes in FS during administration of Nanoxel®-M. (B) Repeated measures ANOVA of changes in FS (p=0.0370). EORTC=European organization for research and treatment of cancer; QLQ=quality of life questionnaire; TC=Nanoxel®-M+cyclophosphamide.
The SS of EORTC QLQ-C30 is based on dyspnea, pain, fatigue, sleep disturbance, appetite loss, nausea, and vomiting. In general, the SS of all three groups showed an increasing trend with an increase in the number of treatment cycles. For patients in the 100 mg dosage subgroup of Group 1, the SS changed from 28.2 at baseline to 28.6, 30.4, 28.8, and 34.5 at cycles 1, 2, 3, and 4, respectively, whereas the SS of patients in the 75 mg dosage subgroup of Group 1 changed from 31.1 at baseline to 26.5, 27.9, 34.9, and 32.1 at cycles 1, 2, 3, and 4, respectively. Similarly, among the patients in Group 2, we recorded an increase in the SS with an increase in the number of treatment cycles, from 24.4 at baseline cycle 1 to 24.1, 25.4, and 31.8 at cycles 2, 3, and 4, respectively (Figure 3A). The mean SS value was highest in the 100 mg dosage subgroup of Group 1 and lowest in Group 2. We observed that there was an overlap of the SS curves at a value of approximately 25 and that the mean value of SS was lowest in Group 2, which was statistically significant (p= 0.0125; Figure 3B).

EORTC QLQ-C30 symptom score (SS) changes of patients treated with Nanoxel®-M. (A) Changes in SS during administration of Nanoxel®-M. (B) Repeated measures ANOVA of changes in SS (p=0.0125). EORTC=European organization for research and treatment of cancer; QLQ=quality of life questionnaire; TC=Nanoxel®-M+cyclophosphamide.
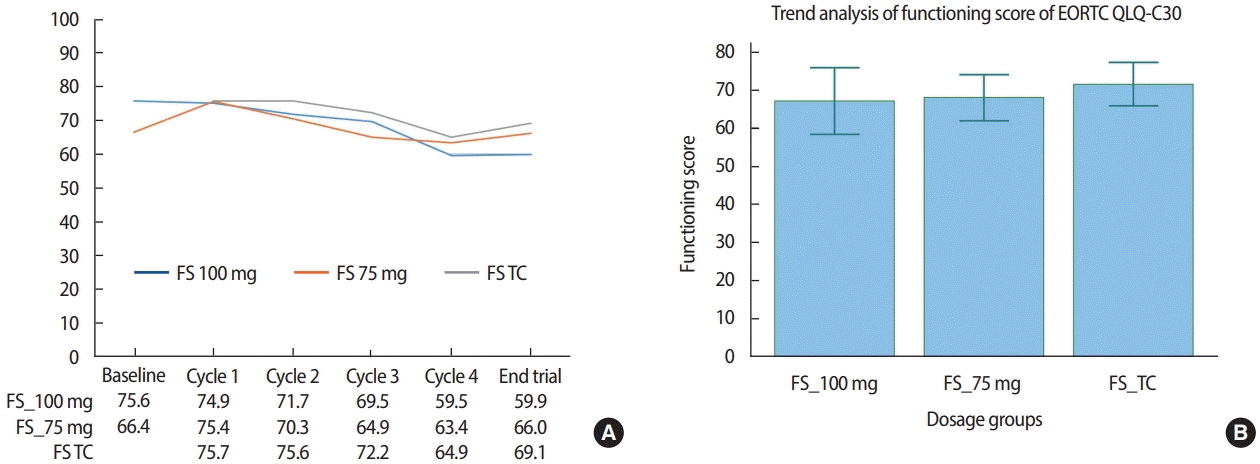
For all three patient groups, we observed a gradual decrease in the GHS of EORTC QLQ-C30 with an increase in the number of treatment cycles. Specifically, the GHS of patients in the 100 mg dosage subgroup of Group 1 changed from 58.3 at baseline to 54.8, 46.3, 52.8, and 43.2 at cycles 1, 2, 3, and 4, respectively, whereas the GHS of patients in the 75 mg dosage subgroup of Group 1 changed from 51.8 at baseline to 68.0, 56.7, 53.5, and 45.7 at cycles 1, 2, 3, and 4, respectively. Similarly, the GHS of Group 2 patients changed from 56.7 at baseline cycle 1 to 55.0, 50.0, 46.7, and 55.0 at cycles 2, 3, and 4, and the end of the trial, respectively (Figure 4A). The mean value of GHS was lowest in the 100 mg dosage subgroup of Group 1 at 48.5, whereas values for the 75 mg dosage subgroup of Group 1 and Group 2 were similar at 53.7 and 52.7, respectively; the difference between these values was not statistically significant (p= 0.3445; Figure 4B).
EORTC QLQ-C30 global health status score (GHS) changes of patients treated with Nanoxel®-M. (A) Changes in GHS during administration of Nanoxel®-M. (B) Repeated measures ANOVA of changes in GHS (p=0.3445). EORTC=European organization for research and treatment of cancer; QLQ=quality of life questionnaire; TC=Nanoxel®-M+cyclophosphamide.
DISCUSSION
Docetaxel has been recommended as a standard therapeutic drug for the treatment of breast cancer in adjuvant and metastatic settings, in conjunction with the administration of anthracyclines [16]. Multiple large-scale, randomized, clinical trials have indicated that the sequential addition of docetaxel to doxorubicin with cyclophosphamide is more effective than dose-dense AC as a preoperative treatment for patients with operable breast cancer [17,18]. The US Oncology Research Trial 9735 demonstrated that four cycles of docetaxel/cyclophosphamide resulted in survival rates superior to those obtained using AC in early breast cancer patients [19]. The frequency of grade 3/4 neutropenia recorded in this trial was 61%; febrile neutropenia was 5% without the administration of granulocyte colony-stimulating factor. Based on the results obtained in the 9735 trial, we recommended that investigators use the TC regimen for patients with the endocrine responsive (ER-positive and/or PR-positive), low proliferative (Ki67 less than 14% and HER-2-negative) luminal A subtype of operable breast cancer, which is a more conservative approach than AC followed by docetaxel.
In terms of the safety and toxicity of Nanoxel®-M, we observed a higher incidence of febrile neutropenia in patients in the 100 mg dosage subgroup of Group 1, which is similar to the incidence of neutropenia reported in the USON 9735 trial [19]. Accordingly, all participating investigators agreed that it would be prudent to reduce the dose of Nanoxel®-M administered to Group 1 patients from 100 mg/m2 to 75 mg/m2, which was approved by each IRB after the enrollment of 18 patients. Following this dose reduction, there was a marked decrease in the incidence of grade 3/4 neutropenia from 81.9% to 56.5%, which patients found tolerable, as indicated by the fact that none of the patients enrolled in the trial subsequently dropped out. Given that a higher incidence of grade 3/4 neutropenia has repeatedly been reported in Asian, rather than Caucasian, women with breast cancer [20-22], it is important for clinicians to consider ethnic differences, which are important factors for improving the treatment outcomes for cancer patients, because chemotherapy-induced toxicity might contribute to lower survival. Accordingly, we realized that a 100 mg/m2 dose of Nanoxel®-M represents an overdose for Asian women with breast cancer.
Among the 220 cycles monitored in the present trial, we recorded only two grade 1 hypersensitivity reactions. A survey of the relevant literature indicated that hypersensitivity reactions occur in approximately 25% to 50% of patients in response to treatment with taxanes, whereas severe anaphylactic reactions have been reported in 2% to 4% of patients. Preventive medications for hypersensitivity reactions routinely includes the administration corticosteroids and antihistamines, which have been found to be effective when used in conjunction with taxanes [23-25]. Compared with previous studies that have examined the effects of paclitaxel and docetaxel, we observed a lower incidence of hypersensitivity in response to treatment with Nanoxel®-M, which appears to reflect the fact that in terms of hypersensitivity reactions, Nanoxel®-M is a safer option than docetaxel used in conjunction with the nonionic surfactant, polysorbate 80 (Tween 80).
Chemotherapy-induced peripheral neuropathy is an important indicator of the need for treatment modification, interruption, or discontinuation [26]. Taxane-induced neurotoxic symptoms are related to paresthesia of the extremities, manifesting as numbness, tingling, and shooting or burning pain [27]. In this regard, Song et al. [28] examined the incidence, risk factors, and prescribing patterns associated with taxane-induced peripheral neuropathy (TIPN) in Korean clinical practice. They reported that the incidence of TIPN in paclitaxel and docetaxel treatment groups was 89.9% and 69.0%, respectively, but 42.2% and 15.8%, respectively, in patients who were administered anti-neuropathic agents during taxane treatment.
In the present study, we found that the average incidence of TIPN in patients treated with Nanoxel®-M for all three treatment groups was approximately 9.1%, which was lower than that reported previously in response to treatment with docetaxel. However, the incidence of TIPN was notably higher (16.7%) in the 100 mg/m2 Nanoxel®-M subgroup of Group 1 patients, which is similar to the incidence previously reported in response to docetaxel. Nevertheless, we observed a decrease in the incidence of TIPN from 16.7% to 5.6% following a reduction in the dose of Nanoxel®-M administered to Group 1 patients. This indicates that in terms of hematological and non-hematological toxicity, a Nanoxel®-M dose of 75 mg/m2 per cycle is more appropriate for Korean women with breast cancer in an adjuvant setting. Furthermore, we should also recognize that TIPN occurs in proportion to the dose level and cumulative dose, and might affect the QoL of breast cancer patients who are treated with taxanes.
Fayers et al. [29] reported that the reference data for the EORTC QLQ-C30 is based on the general population; the members of which have a far-from-perfect QoL. They also recommended that clinicians should strive to minimize the percentage reduction in QoL, as compared with the baseline. When we evaluated the alterations in QoL with respect to the three categories of physical activity, symptomatology, and global health, we observed gradual decreases with minor fluctuations in FS and GHS and an increase in SS in all three treatment groups. Overall, FS and GHS were highest; SS was lowest in the TC group, which indicates that the patients in this group who had received no previous anthracycline treatment maintained the highest level of physical activity and general health, as well as fewer disease symptoms. Additionally, we observed that a reduction in the dose of Nanoxel®-M led to only a minimal reduction in QoL.
This study has some limitations. First, the numbers of patients in each group are relatively small, especially in the 100 mg/m2 Nanoxel®-M subgroup of Group 1. Second, we did not compare the oncologic outcomes between the subgroups of Group 1 because the aims of this study were the safety, toxicity of Nanoxel®-M and the QoL of breast cancer patients.
In conclusion, the findings of this study indicate that, as a taxane, Nanoxel®-M treatment for operable breast cancer patients compares favorably with docetaxel in terms of hypersensitivity reactions and the incidence of TIPN and is associated with a similar incidence of febrile neutropenia. Furthermore, we observed decreases in the hematological and non-hematological toxicities of Nanoxel®-M following a reduction in dose from 100 mg/m2 to 75 mg/m2, thereby indicating that a 75 mg/m2 dose of Nanoxel®-M would be more appropriate than 100 mg/m2 for the QoL of breast cancer patients.
Notes
This clinical trial was sponsored by the Samyang Biopharmaceuticals Co. (Seongnam, Korea). The all authors declare that they have no other conflicts of interests.